

ClonExpress II One Step Cloning Kit
非连接酶依赖型的单片段一步法克隆试剂盒

快速克隆;高通量克隆;无缝克隆;DNA定点突变;双链DNA与载体组装

· 简单、快速、高效,适用于任何载体
· 无需考虑插入片段携带的酶切位点
· 可高效克隆 50 bp-10 kb 的 DNA 片段
· 线性化克隆载体和 PCR 产物可不纯化直接克隆
· 无连接酶,无自连,阳性率 >95%
· 提供在线引物设计软件CE Design
1.克隆效率高
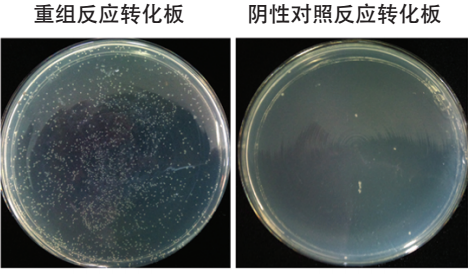
图1
重组产物转化后平板上一般可形成数千个单克隆,而无插入片段的阴性对照反应转化后只有数个单克隆形成。因此ClonExpress 系统的自连背景会大大降低连接酶依赖的克隆方式。
2.克隆阳性率95%以上
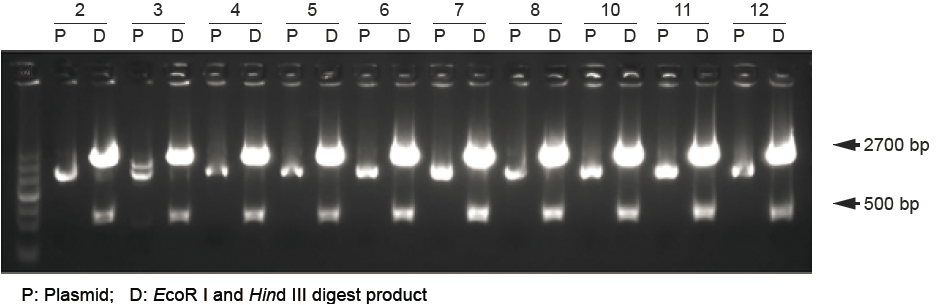
图2
鉴定酶切结果表明,克隆阳性率可达95%以上。

本试剂盒基于简单、快速、高效的DNA无缝克隆技术,可将插入片段定向克隆至任意载体的任意位点的。试剂盒独特的非连接酶依赖体系降低了载体自连背景;高度优化的反应缓冲液及增强的重组酶提高了克隆的重组效率与对杂质的耐受度,仅需反应30 min即可进行转化,完成定向克隆,阳性率可达95%以上。



- 30℃~-15℃保存
C112
Q1:引物如何设计?
引物由三部分组成:同源臂(15-20 bp,不计算酶切位点和残留碱基,GC含量40%-60%)+酶切位点(按需保留或舍弃)+特异性引物(引物Tm值由特异性序列决定,计算不包括同源臂和酶切位点的序列)
- 引物设计:官网引物设计软件CE Design,选择相应模块进行设计。
- 载体的线性化方式有三种:双酶切、单酶切和反向PCR,优先选择双酶切;
Q2:阳性对照不长斑或很少。
- 平板抗性使用错误:C112,C113试剂盒中提供的对照载体的抗性为Amp+抗性,C115对照载体的抗性为Amp+,Kan+双抗性;
- 感受态细胞效率低:确保转化效率>107cfu/μg,可进行简单检测,将1 ng质粒转化至100 μl感受态细胞中,取1/10进行涂板,生长1000个菌斑,估算感受态转化效率为107cfu/μg;重组产物的转化体积不应超过感受态细胞体积的1/10,否则会降低转化效率;感受态选择克隆感受态(如DH5α/XL10),不能用表达感受态。
Q3:平板上长不出克隆/克隆数很少/空载体
- 阳性对照:用于排查是否因为操作原因、感受态效价下降、试剂酶活下降导致的重组效率低。
- 引物设计不正确:引物包含15-20 bp同源臂(不计算酶切位点),GC含量40%-60%;若同源臂的长度小于13bp或者大于35bp,基本没有重组活性,长度大于25bp,重组效率会有明显下降;GC含量过高或过低,会影响重组效率。
- 载体线性化方式:尽量选用双酶切,若为快速酶切,建议延长酶切时间(若无星号活性可以过夜酶切),减少质粒的投入量,采用胶回收进行纯化以降低载体未被切开的概率,也可以做阴性对照来验证载体线性化程度。
- 载体和插入片段未切胶回收:未纯化DNA有可能会抑制重组反应, 建议线性化载体、PCR产物进行凝胶回收纯化,纯化产物溶解在ddH2O中。
- 线性化克隆载体和插入片段浓度测定不准确。如果使用NanoDrop测定浓度,必须保证浓度值大于20ng/μl,同时OD260/280在1.8附近,才能保证得到的浓度较为准确,否则需要跑胶鉴定其浓度或重新制备片段(多管浓缩以增加回收浓度)。
- 线性化克隆载体和插入片段扩增产物的使用量不足/过量,即比例不佳:严格按照说明书推荐的量和比例配制重组体系,各组分添加不低于1μl(避免低体积添加造成量取不准)。
- 重组体系:对于C112/C113,重组体系为20μl,10μl重组体系容易造成重组效率低。
- 重组条件:反应需要在精密仪器中进行,如PCR仪、金属浴等,不建议在水浴锅、恒温箱中进行重组,后者可能因为温度不准确造成重组效率下降。
- 感受态细胞的转化效率需大于107cfu/μg,重组产物的转化体积不应超过感受态细胞体积的1/10,否则会降低转化效率;选择克隆用感受态细胞(如DH5α/XL10),不能选择表达用感受态细胞。若为自制的感受态细胞需要测定效价(将1ng质粒转化至100μl感受态细胞中,取1/10进行涂板,生长1000个菌斑,估算感受态转化效率为107cfu/μg),且涂板时需将感受态细胞全部涂在平板上;感受态转化温度准确。
- 增加重组效率的辅助手段:对于C112可以等比例减半载体与片段的投入量。
- 除此之外,需要检查抗生素抗性有未加错。
Q4:假阳性/测序无信号/空载体
- 阴性对照:用于排查是否是质粒载体残留导致的假阳性,如果阴性对照长了很多,可能是模板投入量过多或者未进行DpnI消化。
- 以质粒为模板PCR获得片段/载体:质粒投入量不宜过多(<1ng),否则可能导致DpnI消化不完全从而造成假阳性。建议PCR产物进行凝胶回收纯化,纯化产物溶解在pH 8.0的ddH2O中。
- 挑斑方法:建议选取平板上某一块区域,同时挑选大斑和小斑。主要原因是有以下几点:重组产物3'端与5'端未连接,需要到感受态细胞中进行修复,斑点相对来说会小点;另外板子营养不均匀会导致斑点大小有差异;重组质粒相对原始质粒来说,有可能会影响细胞的生长。所以建议挑取大小斑点,以增加挑到重组产物的概率。
- 菌检引物:建议使用载体通用引物或者引物一端在载体上,一端在目的片段上。如果用目的片段引物进行菌液PCR,可能会出现菌检有结果而测序无结果。
Q5:菌液PCR无条带
⑴ PCR体系或程序不合适:没有目的条带也没有空质粒条带,建议优化PCR体系、程序;或者提取质粒,以质粒做模板PCR验证,或进行酶切验证。
⑵ 重组失败:只有空质粒的条带,说明重组不成功,载体线性化不完全,建议优化酶切体系,检查引物设计。
⑶ PCR试剂不合适:试剂不适合粗品,或者是超出了试剂的使用范围,扩增的片段太大。
Q6:多数克隆含有不正确的插入片段
⑴ PCR产物混有非特异性产物:优化PCR体系,提高特异性;胶回收PCR产物;鉴定更多的克隆。
⑵ 反应体系中混入了相同抗性的质粒:PCR扩增模板为环状质粒时,如扩增产物直接用于重组反应时需进行DpnI消化,或者进行胶回收纯化。
Q7:插入片段出现碱基突变
- 测序的样本数:如果只测了一个样本,不一定可信,需要多测几个样本。
- 分析测序结果:检查突变处的测序信号是否有问题,若为双峰或者杂峰则不一定可信。
- 检查突变位点:如果多个结果突变位点一致,可能是从模板上的引入的或者模板序列与NCBI上不一致。
- 插入片段在PCR获得时使用高保真的聚合酶。
- 若引物处发生碱基突变,考虑是引物合成问题,建议重新合成引物进行实验。
相关产品
GR501-01/02
FastPure Gel DNA Extraction Mini Kit
DC301-01
2 × Phanta Flash Master Mix(Dye Plus)
P520-01/02/03
C505-02/03
P222-01/02/03/04
DC201-01



